分享到:
- 微信
- 微博
分享到微信打开微信,点击底部的“发现”, |



{{aisd}}
AI生成 免责声明
作为创新药加快上市的四大“绿色通道”之一,“药品附条件批准上市”允许未完成全周期临床试验的药品有条件上市,显著缩短了临床急需药物的可及时间。然而,新药上市前“疗效未充分验证”“长期安全性缺失”,上市后确证性研究缺失、持有人变更等因素,也客观上造成了新的药品安全隐患。
近年来,一些药企借由附条件批准上市通道将创新药快速推向市场,但拖延多年未能完成确证性研究,以至将“附条件上市”的“临时证书”作为药品上市流通的“长期凭证”。
与此同时,随着更多附条件批准上市的新药纳入医保,相关赛道的药品研发竞速,医保基金和产业研发资金的使用效益也可能因此受到冲击。
近日,国家药监局正式发布《药品附条件批准上市申请审评审批工作程序》(下称《工作程序》)。在试行五年多时间,历经两次征求意见后,正式版文件将“确证性研究”的监管贯穿药品附条件上市“审批—执行—退出”始终。国家药监局明确,附条件批准的确证性研究原则上需在上市后4年内完成,延期原则上仅有一次且药品注册证书有效期不随延期延长,这意味着药企“只上车不补票”将成为过去式。
“同质化创新”试图“搭便车”的行为也将终止——若已有药品获得常规批准,其他在研或在审的“同机制、同靶点、同适应证”药品的附条件批准通道将关闭。“严控附条件审评标准,严防泛化。”一名接近CDE(国家药监局药品审评中心)的专家如是说。
“上市后补临床”原则上不超过4年
随着《工作程序》落地,附条件批准上市的新药将自上市之日起,就进入药品注册证书有效期调整的“倒计时”阶段——要么4年内完成确证性研究,转为常规申请并开展药品再注册工作;要么注销药品注册证书或核减适应证,药品“退市”。
附条件批准上市,是指用于严重危及生命且尚无有效治疗手段的疾病、公共卫生方面急需的药品,现有临床研究资料尚未满足常规上市注册的全部要求,但已有临床试验数据显示疗效并能预测其临床价值,在规定申请人必须履行特定条件的情况下基于替代终点、中间临床终点或早期临床试验数据而批准上市。
2020年修订的《药品注册管理办法》确定了四种药品加快上市注册程序,包括突破性治疗、附条件批准、优先审评审批和特别审批。同年,国家药监局发布《药品附条件批准上市申请审评审批工作程序(试行)》。
相较于其他三种加速许可程序,附条件批准上市之所以备受产业界关注,是因为它可显著缩短药物临床试验时间,为热门赛道的新药率先抢占市场争取时间。
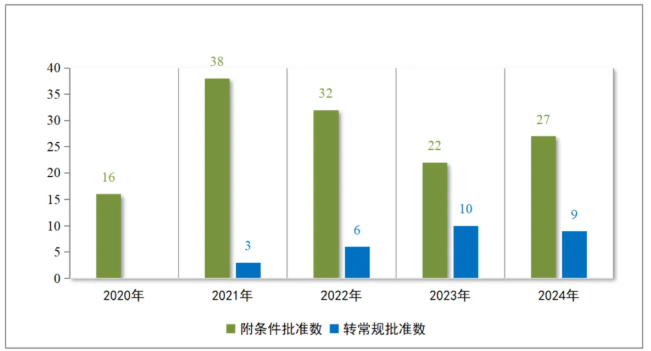
国家药监局去年在《2024年度药品审评报告》中披露了一组数据:自2020年《药品注册管理办法》实施以来,共有187件药品注册申请(135项适应证)附条件批准上市。
但在获得附条件批准上市后,药品上市许可持有人还需按照药品注册证书中所附的特定条件,开展新的或继续正在进行的临床试验,这些临床试验通常是以确认预期的临床获益为目的的确证性临床试验,为常规上市提供充足证据。
“确证性临床试验”的完成率是附条件批准上市制度最为业界关注的风险之一,事关患者用药安全。根据前述药审报告,自2020年以来,仅有28项附条件批准的适应证完成确证性研究,转为常规批准。
第一财经梳理国家药监局药品审评中心(CDE)官网“附条件批准品种”还发现,截至4月26日,在已经附条件批准上市的药品中,有相当比例药品上市后确证性研究的既定年限为4~5年;部分在2020~2022年附条件批准上市药品中并未明确药品注册证书有效期时间;部分于2020年附条件上市的新药,今年药品注册证将到期,但上市后确证性研究至今尚未完成;此外也有少数附条件上市药品规划用6年时间完成确证性研究时间,这已超过了常规药品注册证书有效期年限(5年)。
为减轻附条件批准药品的有效性风险,减少临床医生使用顾虑,《工作程序》从上市申请前沟通、上市后监管到常规批准转换或药品注册证书注销,全流程加强确证性研究监管。
附条件批准上市前,药品确证性研究理论上应已启动。根据《工作程序》,确证性研究的方案和完成时限应由药审中心在审评中与申请人沟通交流后确定。若直至审评结束,CDE仍未收到药企启动确证性研究证明材料,将作出不通过的审评结论。
同时,上市后的确证性研究不再“无限期延长”。《工作程序》明确,自附条件批准上市之日起,确证性研究原则上不超过4年。每年度,药品上市许可持有人还需向CDE报告确证性研究进展,随安全性更新报告一并提交。
确证性研究时长与药品流通销售深度挂钩——附条件批准时,每个附条件批准的适应证单独设置药品注册证书有效期,原则上,在确证性研究完成时限的基础上增加一年。在“已完成全部确证性研究,且提交的全部数据可以证明获益大于风险”的情况下,药品上市许可持有人可调整药品注册证书有效期,申报转为常规批准的注册申请。
相反,如果综合原有研究和药品上市许可持有人新提交的研究资料不能证明其获益大于风险的,药企则会面临药品注册证书注销或适应证核减。此外,如果药品上市许可持有人终止/完成确证性研究,自行评估其研究结果无法确证药品安全有效性,也应当立即主动停止相关适应证的药品销售。
有从事药品临床研发的受访人士认为,这些新规相当于对药品上市许可持有人“从快完成确证性研究”提出制度性激励和制度性约束,尤其是“药品注册证书有效期不予延长”的规定,有望显著增加持有人及早完成上市后研究的主动性。
不过,以罕见病用药为代表的新药研发,客观上存在受试者招募困难等堵点。针对新药附条件批准上市后,临床试验进展不顺,无法如期完善既定规划或者需要更换研究方案等情况,《工作程序》也给出申请延期通道。但申请延期的补充申请原则上不超过一次,且药品注册证书有效期保持不变。
附条件申请、仿制研发通道收紧
随着国内多赛道新药研发“内卷”加剧,一款新药附条件批准上市后,同类药品能否开展相似的以附条件上市为目标的临床试验申请?相关仿制药或生物类似药何时可以开展临床和申报上市?这些问题的答案,牵动产业界的神经。
为了让有限的优先审评审批资源“用在刀刃上”,避免热门管线研发扎堆和同质化,根据《工作程序》,已有药品获得常规批准后,其他在研或在审的同类药品不再符合附条件批准的要求。
《工作程序》同时提到,附条件批准上市的药品,在其转为常规批准之前,原则上,该品种不发布为参比制剂。
具体来说,对于附条件批准上市的化学药品,在其转为常规批准并发布为参比制剂之前,申请人可以开展相关仿制研究工作,但不受理该品种仿制药上市注册申请;对于生物制品,在我国附条件批准上市的生物制品在其转为常规批准之后,方可受理该品种生物类似药上市申请。
参比制剂是仿制药研发和评价的“标准参照物”。不发布参比制剂,不受理相关仿制药或生物类似药的上市申请,在业界看来,一方面意味着附条件批准上市的新药有望在一定时间内获得市场独占期,另一方面也是因为此类新药的疗效与安全仍待确证,需要从源头上避免仿制药企依据不确定的证据,浪费研发资源。
如需获得授权请联系第一财经版权部:banquan@yicai.com